Congenital long QT syndrome: A case report
Resident Internal Medicine, Jersey City Medical Center, NJ, USA

|
Case Report
Congenital long QT syndrome: A case report
Resident Internal Medicine, Jersey City Medical Center, NJ, USA
|
|
Abstract
The congenital long QT syndrome (LQTS) is characterized by abnormally prolonged ventricular repolarization
due to inherited defects in cardiac sodium and potassium channels, which predispose the patients to syncope, seizure
like activity, ventricular arrhythmias, and sudden cardiac death. Early diagnosis and preventive treatment are
instrumental in preventing sudden cardiac deaths in patients with the congenital LQTS. The diagnostic criteria
for congenital LQTS are based on certain electrocardiographic findings, clinical findings and findings of epinephrine
stress test. Recently genotype specific electrocardiographic pattern in the congenital LQTS has also been
described. Recent studies suggest feasibility of genotype specific treatment of LQTS and, in near future, mutation
specific treatment will probably become a novel approach to this potentially fatal syndrome. We describe one case
that fulfilled the electrocardiographic, historical diagnostic criteria and epinephrine stress test suggestive of LQT
syndrome.
Key words
congenital long QT syndrome; cardiac sodium; potassium channels
J Thorac Dis 2010;2:185-188. DOI: 10.3978/j.issn.2072-1439.2010.02.03.12
|
|
Introduction
The Long QT is a rare congenital disorder characterized by
QT-interval prolongation and repetitive episodes of syncope
and cardiac arrest related to rapid, polymorphic ventricular
tachycardia. Genetic linkage mapping defines six types of LQTS
(LQT1-LQT6) out of which, LQT1-LQT3 have been well
characterized in clinical studies (1). Diagnosis of LQTS is based
on clinical and electrocardiographic features (2). These EKG
characteristics are useful for selecting which gene to investigate
first, while performing genetic analysis. The identification
of genotype specific EKG pattern is gaining importance, for
its potential use in the management of LQTS, with favorable
outcomes (3). Further, epinephrine stress test is important in
unrevealing the underlying congenital LQTS.
|
|
Case report
A 29 year-old Caucasian male, recently diagnosed with seizure
disorder, was brought to emergency room for altered mental status with combative behavior. Patient was having syncopal
episodes and sudden seizures with spontaneous resolution in few
minutes for the past 4-5 months. Family history was significant
for sudden deaths in family in early age.
In ER, the Patient was initially found to be combative but
hemodynamically stable. 12 lead EKG showed presence of long
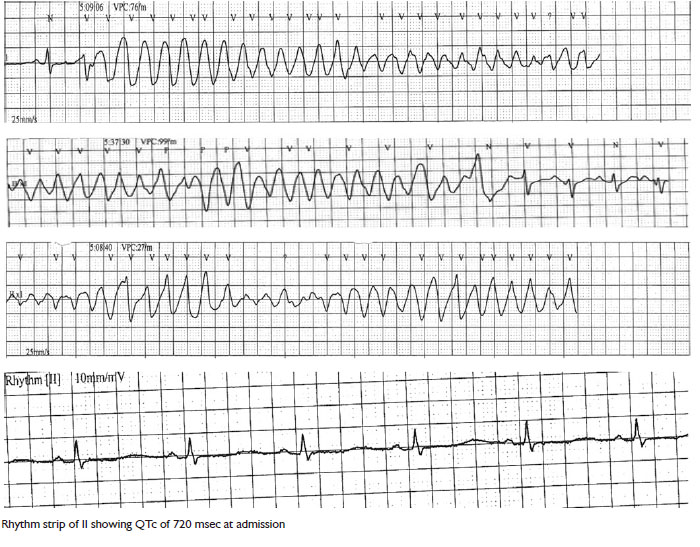
corrected QT interval of 710 msec, and presence of U waves
correlating with his low potassium levels of 3.1. The urine
toxicology screen was positive for cocaine. Patient’s mental
status rapidly improved and in a span of few hours he was able
to provide coherent history. Suddenly, he was found to have a
seizure-like activity in the ER. Monitor revealed a wide complex,
polymorphic ventricular tachycardia, which reverted to sinus
rhythm in a span of few seconds. After the initial episode, patient
had 2 more episodes of ventricular tachycardia in the next
few minutes, which resolved spontaneously after lasting for a
duration of 5-7 seconds.
Patient was initially intubated and transferred to Cardiac
Care Unit (CCU). Serial EKG’s done in the CCU did reveal a
prolonged QTC which gradually decreased to normal duration
over a span of 2 days. Given the history of sudden cardiac deaths
in the family and seizure like episodes in the patient, diagnosis
of Congenital long QT syndrome was entertained and an
Epinephrine induced QT stress test was planned.
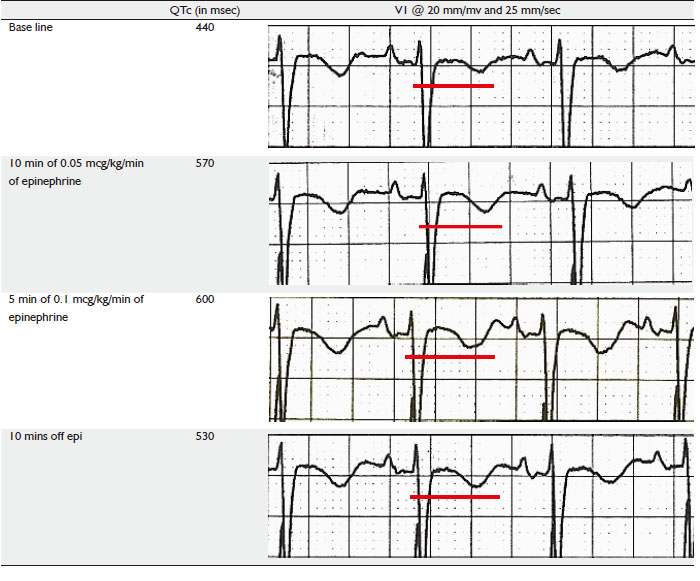
Initial EKG before the start of epinephrine stress test
showed a QTc of 440 msec, patient was started on Epinephrine
infusion at rate of 0.25 micrograms/Kg/min for 10 mins under
continuous EKG monitoring. QTc at end of this period did show
a prolongation to 570 msec after which rate of epinephrine was increased to 0.5 mcg/kg/min for another 5 minutes, at end of
which QTc prolonged to 600 msec. The test was deemed positive
and was stopped at this point of time. Patients QTc rapidly
reverted to pre test level after epinephrine was stopped.
Patient was given an External defibrillator vest and discharged
home with close outpatient follow up and plan for repetition of
QTc stress test in 2 months time for evaluation of need for AICD.
  |
|
Discussion
The QT interval is a surface marker of cardiac electrical activity,
specifically cellular repolarization. It is generally accepted that
the absolute QT interval provides a surface rendering of the
underlying cellular action potential durations. Despite overlap
of the resting QTc between healthy persons and patients with
LQTS, the 12-lead EKG remains one of the principal tools in the
LQTS evaluation, and the baseline QTc is still one of the most
important diagnostic criteria.
The congenital LQTS is a potentially life threatening condition, caused by mutations in genes encoding cardiac ion
channels which result in prolongation of ventricular action
potential. Genetic screening of symptomatic patients or their
asymptomatic family members may identify patients at risk
for life threatening arrhythmias and the type of LQT as it has
important implications in the management. Out of the several
forms of congenital LQTS, three forms LQT1, LQT2, and
LQT3 have been well characterized. These three forms have also
been described on the basis of their specific EKG morphology.
Recent investigations suggest that even in patients with acquired
LQTS (e.g. resulting from intake of QT-prolonging medicines),
there are clinically silent gene mutations that lead to overt QT
prolongation only with exposure to QT- prolonging medications
(4-6).This explains why some patients seem to be more prone
than others to have QT prolongation at a given dose of QTProlonging
drugs, even after adjustment for other factors that
could prolong QT-interval.
According to Pfizer Tikosyn program, the QTc should be no
more than 500 msec in the presence of ventricular conduction abnormality (7). This guidance may be used until a standard
method is established for the measurement. Treatment with
beta-blockers can reduce this risk. The clinical course of the
congenital LQTS is influenced largely by the gene affected (8).
While cardiac events are more frequent and occur at a younger
age in patients with LQT1 and LQT2, they are potentially more
fatal in patients with genotype LQT3. Patients with LQT1 and
LQT2 genotype typically benefit from high dose beta-blocker
therapy (9, 10). However, patients with LQT3 are at higher
risk at lower heart rates and potentially may benefit from pace
maker therapy. In addition, they shorten their QT-interval more
with sodium channel blockers (11).
Provocative tests using catecholamine or exercise testing have
long been considered to unmask some forms of congenital LQTS
(12). Recent preliminary data by Ackerman et al. have suggested
the usefulness of an epinephrine test to unveil concealed LQT1
syndrome (13). An epinephrine provocative test should only be done by cardiologists, under enough preparation of intravenous
beta-blockers and direct cardioverter for unintentionally induced
ventricular fibrillation.
Both experimental and clinical studies have suggested a
differential response of action potential duration (APD) and QT
interval to sympathetic stimulation among LQT1, LQT2, and
LQT3 (14). Persistent and paradoxical prolongation of APD
and QT interval at steady state conditions of catecholamines is
reported in LQT1 syndrome. Under normal conditions, betaadrenergic
stimulation is expected to increase net outward repolarizing
current, owing to larger increase of outward currents,
including Ca-activated slow component of the delayed rectifier
potassium current (IKs) and Ca-activated chloride current, than
that of an inward current, Na/Ca exchange current (INa-Ca),
resulting in an abbreviation of APD and QT interval. A defect
in IKs in the LQT1 syndrome could account for failure of betaadrenergic
stimulation to abbreviate APD and QT interval, resulting in a persistent and paradoxical QT prolongation
under sympathetic stimulation (14). In LQT2 syndrome,
catecholamines are reported to initially prolong but then
abbreviate APD and QT interval, probably because of an initial
augmentation of INa-Ca and a subsequent stimulation of IKs.
In contrast to the LQT1 and LQT2 syndromes, catecholamines
are reported to constantly abbreviate APD and QT interval as
a result of a stimulation of IKs in the LQT3 syndrome, because
an inward late sodium current (INa) was augmented in this
genotype. The epinephrine test may be applied not only for
unmasking silent mutation carriers with LQT1 syndrome but
also for predicting genotypes.
Facilities for genetic analysis are not easily available. However,
in view of the growing importance of genotype specific
treatment of this potentially fatal syndrome, one can utilize
the ECG criteria and epinephriene QT stress test as a reliable
indicator of the underlying genotype and accordingly tailor the
management.
|
|
References
Cite this article as: Aziz F, Penupolu S, Doddi S, Togonu-Bickersteth B, Ameen A. Congenital long QT syndrome: A case report. J Thorac Dis 2010;2(3):185-188. doi: 10.3978/j.issn.2072-1439.2010.02.03.12
|

