In the past four decades, respiration during sleep has intrigued clinicians and basic scientists, probably owing to a better recognition of the clinical significance of sleep apnea syndrome. Sleep apnea syndrome is characterized by repetitive episodes of cessation of airflow at the nose and mouth lasting at least 10 seconds during sleep in association with one or more of the following symptoms: habitual snoring, restless sleep, morning headache, excessive daytime sleepiness and intellectual impairment (
1). Although the phenomenon of sleep disordered breathing (SDB) has long been noticed (
2,
3), the first polysomnographic recording of frequent respiratory pauses during sleep in humans dates back to 1965 (
4).
As the term suggests, sleep apnea involves two control systems, i.e., those controlling consciousness and respiration. What interests us is that recurrent apneas occur predominantly, if not entirely, during sleep. This phenomenon raised a fundamental question: how the sleeping state itself can permit—or even provoke—apneas in an otherwise healthy respiratory system, which is precise and mechanically efficient in wakefulness?
It has long been known that sleep generally depresses respiration, circulation and other vital activity mainly due to the reduced metabolic rate and sympathetic activity. However, SDB reveals more complex effects of sleep on ventilation than merely depression. In general, sleep may disturb breathing via the following mechanisms: 1) functional changes in the central nervous system, such as the loss of the wakefulness stimulus; 2) the state related fluctuation of excitatory and inhibitory impact on respiration; 3) decline of skeletal muscle tone; 4) attenuation of ventilatory response to chemical and mechanical loads; and 5) reduction in functional residual capacity (FRC) and mismatch of ventilation/blood (
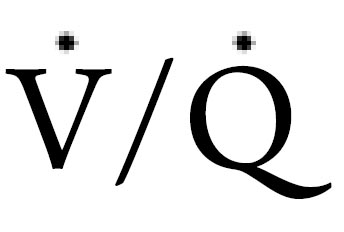
) ratio related to recumbent position. The follow discussion will examine the implications of the aforementioned factors on breathing stability and respiratory pattern during sleep.
Loss of wakefulness drive to breathing control
Like all oscillators, the respiratory rhythm generator requires tonic inputs to keep oscillations between inspiration and expiration, which is mainly provided by chemical drive and wakefulness stimulus. The wakefulness stimulus, a non-specific drive, might arise from suprapontine regions or the reticular activating system (
5). During wakefulness, the tonic input from the wakefulness stimulus to the respiratory center is sufficient to compensate for reductions in chemical stimuli and sufficient to overcome other inhibitory factors, so that apnea
rarely occurs in awake humans even in the presence of substantial
hypocapnia such as vigorous and long-lasting singing or crying.
In contrast, a sleep-related loss of the wakefulness stimulus leaves
ventilation under metabolic control, making the respiration
control system very sensitive to any transient reduction of
PaCO
2 and predisposing those sleepers with low CO
2 reserve or
high upper airway collapsibility to apnea. (
6,
7). Furthermore,
the loss of wakefulness stimulus, coupled with the reduced
respiratory muscular tone during sleep may exacerbate alveolar
hypoventilation in some diseases, making marginal ventilation
during wakefulness become inadequate during sleep. Hence, the
withdrawal of the wakefulness stimulus has the most important
impact on ventilatory control and probably plays a critical role in
the pathogenesis of SDB (
5).
Neurophysiologic changes with sleep state modifies
respiration
Sleep is not a homogeneous state and can be subdivided into
two distinct neurophysiological states based on behavioral and
electrographic characteristics: rapid eye movement (REM) and
non-rapid eye movement (NREM) sleep. Dynamic changes of
sleep state with alterations in the degree of alertness are also
potential factors in destabilizing breathing.
Slow wave NREM sleep is featured by the stability of
autonomic regulation with an absent waking neural drive
and a quiescent behavioral system (
8). Therefore, respiration
is relatively stable in slow wave sleep (
9). In contrast, light
sleep stages, especially stage I sleep and transition periods are
characterized by unstable autonomic regulation. The alpha/
theta-state-specific alterations may lead to ventilatory instability
at or even before sleep onset (
10,
11).
Rapid eye movement sleep is constituent of tonic
motor inhibition and bursts of phasic events. In the tonic
phase, breathing is still under chemical-metabolic control as it
is in NREM sleep. However, in the phasic phase, the breathing
pattern is mainly affected by the behavioral system through the
REM sleep processes, fluctuating with ponto-geniculo-occipital
(PGO) driven excitatory and inhibitory influences (
5,
12).
Although breathing is often irregular in REM sleep, central
apneas are rarely seen in this stage. That is probably because the
apneic threshold for hypocapnia is masked by non-chemical
PGO-related inputs during REM sleep (
13). Indeed, periodic
breathing in REM sleep is mainly composed of obstructive
apneas and hypopneas. Recent research suggests that REMrelated
withdrawal of excitatory noradrenergic and serotonergic
inputs to upper airway (UAW) motoneurons may reduce
pharyngeal muscle activity, predisposing to OSA (
14).
Moreover, sleep is fragmented by arousals, awakenings and
state-transitions. All of these events may affect breathing stability
(
15). Arousal, a state of heightened brain stem activity, is a
brief awakening (
3-
15 seconds) from sleep induced by various
external or internal influences, including chemical as well as
mechanical stimuli arising from respiratory effort. This protective
mechanism is often triggered by apneas and hypopneas,
helping to terminate them. On other hand, the transition from
sleep to arousal provides excitatory drives to the respiratory
system, enhancing chemoresponsiveness and causing a surge of
ventilation and transient hypocapnea (
16). These changes, in
turn, tend to destabilize breathing. Consequently, arousal often
plays dual roles in SDB as it terminates the existing apnea, but
also triggers new ones.
It appears paradoxical that sleep predisposes patients to
SDB via removal of the wakefulness stimulus, while arousal
facilitates SDB although it restores the wakefulness stimulus.
The explanation for this paradox lies in the transient nature of
arousals and the short-lived nature of this state-related excitatory
respiratory drive (
16). When transient arousal yields to sleep,
the quick abolition of excitatory input makes the respiratory
control system very sensitive to the transient hypocapnia that
results from prior arousal-provoked hyperpnea. This effect
is exaggerated when arousal occurs at the end of apneas or
hypopneas. The strong chemical stimuli built up during apnea,
plus the sudden release of upper airway resistance (R
UAW),
enhances the arousal-provoked hyperpnea, driving PaCO
2 even
lower. Therefore, frequent transitions between sleep and arousal/
wakefulness exaggerates breathing instability.
What is more, ventilation and diaphragm activities show some
dynamic changes throughout the night, and ventilatory response
to CO
2 is subject to modulation by the circadian rhythm (
17).
Further investigation is needed to figure out how the circadian
rhythm and sleep state interact to affect breathing when one is
superimposed on the other.
Reduction of respiratory muscle tone
There is usually a progressive fall in the activity of skeletal
muscles with both respiratory and nonrespiratory functions
from wakefulness to NREM to REM sleep. The motor neurons
are only slightly hyperpolarized during NREM sleep, but this
change becomes more substantial during REM sleep. Therefore,
a marked motor inhibition prevails during REM sleep. Likewise,
postsynaptic inhibition occurs during REM sleep, which is
also responsible for the REM-related hypotonia/atonia of the
somatic musculature (
18). More interestingly, the influence
of sleep on the chest wall muscles and UAW muscles does
not appear to be uniform or parallel. The UAW dilator muscle
activity demonstrates a progressive depression during the night.
As a result, RUAW undergoes a small increase in the early sleep
period but continues to rise with the deepening of sleep (
19). In
contrast, the principal inspiratory muscles are relatively spared from the direct inhibitory influence of NREM sleep (
20). For
instance, diaphragm electromyographic (EMGdi) activity is
only reduced at the onset of sleep with the transition from alpha
to theta (
19), and then gradually recovers to the same or an
even slightly higher level than the resting awake values during
stable NREM (
21). The recruitment of the chest wall muscle
activity as sleep progresses may at least partly result from the
enhanced chemosensory stimulation of CO
2 retention in the
face of increased R
UAW. However, EMGdi often undergoes
intermittent and brief inhibition and fractionation coincident
with PGO waves or eye movements (
22), causing a reduction of
airflow during REM sleep.
Reduced protective reflexes and compensatory
mechanisms
The sleep state imposes a variety of loads on the respiratory
system, for instance, the increased airflow resistance and
increased PaCO
2. Unfortunately, the ability of the respiratory
control system to compensate for chemical and/or mechanical
loads declines, and the response thresholds for many modalities
of stimulation increases during sleep (
23). Among them, the
reduction of protective reflexes involving pharyngeal muscle
dilators has more clinical significance because it may cause
pharyngeal collapse. Moreover, the low sympathetic neural
tone in sleep may desensitize the carotid bodies, reducing
the chemosensitivity to CO
2 and hypoxia. The reduced
compensation ability for these spontaneously occurring
chemical/mechanical loads, in turn, permits UAW occlusion and
CO
2 retention.
Position related alterations in lung function
As one assumes the supine position for sleep, the lung volume,
especially FRC, decreases slightly in healthy humans, but
largely in obese individuals and asthma patients (
24), probably
because of diaphragm cephalad displacement resulting from the
abdomen content pressure. During deep sleep and REM sleep,
respiratory muscle hypotonia, relative hypoventilation and the
low lung compliance will further reduce end-expiratory lung
volume. These postural related and sleep-associated reductions
in FRC would decrease pharyngeal airway caliber and predispose
the UAW to collapse (
25). Moreover, distribution of air and
blood within the lungs changes in the supine posture, worsening
the
mismatch, probably due to atelectasis and less uniform
distribution of air and blood in the lung bases. This effect is more
significant in obese subjects. The reduced gas exchange and gas
storage in supine position likely disturb breathing by increasing
the plant gain (i.e. PaCO
2 response to alveolar ventilation)
and contribute to the rapid fall in SaO
2 during apneas in obese
patients.
The combination and interaction of the above changes
during sleep introduce the following alterations in ventilatory
parameters: i) UAW impedance and total airway resistance
could increase more than twofold compared to the resting
wakefulness (
26) whereas no alteration of the elastic properties
of the respiratory apparatus occurs. If these normal changes in
UAW mechanics are exaggerated by any other factors, then the
airflow limitation, snoring or OSA may occur; ii) A 10-15% of
reduction in minute ventilation, mainly through a decrease in
tidal volume as a result of respiratory muscle hypotonia, often
causes an increase in PaCO
2 and slight oxygen desaturation,
regardless of the reduction in the metabolic rate with sleep state
(
18); iii) under the background of a relatively low ventilation,
there are several sighs (large tidal volume) sporadically occurring
during sleep. These sighs help to open collapsed alveoli but often
trigger periodic breathing through the post-hyperventilation
hypocapnia (
9); iv) ventilatory response to CO
2 is attenuated
during NREM and tonic REM sleep, which allows PaCO
2 to rise
to a small extent during sleep.
In conclusion, sleep has a significant impact on breathing
mainly through withdrawal of the wakefulness stimulus;
alteration of chemical/non-chemical responses; and reduction
of muscular tone, lung volumes, and metabolic rate. Even so,
the effect of sleep on breathing is usually of minor physiological
consequences in most healthy human subjects, and only
predisposes those with compromised UAW patency or
inappropriate ventilatory control systems to SDB (
7).


