T790M and acquired resistance of EGFR TKI: a literature review of clinical reports
Department of Respiratory Medicine, Jinling Hospital, Nanjing University School of Medicine, Nanjing, China

|
Review Article
T790M and acquired resistance of EGFR TKI: a literature review of clinical reports
Department of Respiratory Medicine, Jinling Hospital, Nanjing University School of Medicine, Nanjing, China
|
|
Abstract
Epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) such as gefitinib and erlotinib are promising therapies for patients with advanced non-small-cell lung cancer (NSCLC). Patients with somatic activating mutations in the EGFR gene have dramatic response initially, but would eventually develop resistance to these TKIs. Subsequent studies found that a secondary mutation in the EGFR gene (T790M mutation) and amplification of the MET proto-oncogene could be the main resistance mechanisms involved. The current review is focused on T790M, which is thought to cause steric hindrance and impair the binding of gefitinib/erlotinib. The T790M is present as a minor allele before TKI therapy and accounts for about half of the acquired resistant cases. Conflicting results were reported for gefitinib-resistant, T790M-acquired patients who had switched to erlotinib treatment, which was proposed to be efficacious. The switch therapy was presumed to work for EGFR wild type patients and previously gefitinib responding patients. MET amplification accounts for about 20% of TKI acquired-resistant patients by a different molecular pathway from T790M; some of these patients will also concurrently have T790M mutation and might still not respond to irreversible TKI. As for the detection of T790M, polymerase chain reaction (PCR), especially mutant-enriched PCR was found to be more sensitive than direct DNA sequencing. In addition, whole genome amplification might also be useful and can be incorporated with future noninvasive method for detecting T790M. A better understanding of the mechanisms leading to TKI resistance is crucial in the development of effective treatment and the design of future clinical studies.
Key words
T790M; lung cancer; acquired resistance; TKI
J Thorac Dis 2011;3:10-18. DOI: 10.3978/j.issn.2072-1439.2010.12.02
|
|
Introduction
Epidermal growth factor (EGF) pathway inhibition is now established as an option for first-, second- and third-line treatments of non-small-cell lung cancer (NSCLC). About 10%-20% of NSCLC patients have been reported to respond positively to gefitinib and erlotinib, which are tyrosine kinase inhibitors (TKI) of the EGF receptor (EGFR). The clinical responses to TKI were shown to be associated with EGFR mutations and KRAS mutation. EGFR mutations, predominantly in exons 19 and 21, confer sensitivity to TKI, while KRAS mutation leads to primary resistance. However, despite an initial response to the treatment of EGFR-TKI in responsive patients, most of them inevitably acquire resistance after a progression-free period of about 10 months (1).
The elucidation of the mechanisms underlying acquired TKI resistance is an important issue of clinical relevance. In this regard, mechanisms such as T790M mutation, MET amplification, over-expression of HGF, activation of IGF1R and other factors have been reported to be associated with acquired resistance to EGFR-TKIs (2). Therefore, the ability to detect relevant biomarkers of acquired resistance is critical in maximizing the benefits of TKI therapy.
Among the resistance causing factors, the secondary point mutation T790M, which substitutes methionine for threonine at amino acid position 790 of EGFR gene domain, might play the most important role. Previous reports on T790M showed that this amino acid substitution was due to a C-to-T base pair change at the second letter of codon790 of EGFR (3). The introduction of vectors carrying T790M to cells also caused gefitinib resistance, indicating that T790M causes sensitive tumors to become resistant to EGFR tyrosine kinase inhibitors (2,4). Furthermore, most clinical reports indicated that T790M accounted for half of the acquired resistant TKI cases (5).
Most of the studies on T790M in NSCLC were small, retrospective and heterogeneous. Moreover, the presence of T790M mutation before treatment, the mechanisms of T790M mutation and the detection methodology were still being debated. Therefore, we extensively reviewed the current literature on the role of T790M as a biological marker for TKI acquired resistance.
We hypothesized that T790M might account for majority of TKI acquired resistant cases and the relationship between T790M and MET amplification was intertwined. The objectives of this review was to find out the role of T790M as a biological marker in TKI acquired resistant cases; check whether the switch therapy from gefitinib to erlotinib was useful for the T790M-acquired patients; explore the relationship between T790M and MET amplification, which is another mechanism reported to be related to acquired resistance; and to compare the efficacy of different detective methods of T790M in clinical reports.
|
|
Materials and methods
A PubMed literature review for all studies published in English from Jan 1, 2005 to May 31, 2010 was performed by using the following medical subject heading terms: “T790M and lung cancer”, “T790M and MET’, “TKI acquired resistance and lung cancer”. The inclusion criteria were as follows: clinical reports including case reports related to T790M mutation in lung primary cancers. The data of the samples’ numbers and T790M-positive samples’ numbers were indispensable for our analysis. The exclusion criteria included English articles, reviews or reports of other primary cancers, basic researches (both in vivo and in vitro), reports whose treatment used other TKIs without gefitinib and erlotinib; articles which mainly talked about other mechanisms of acquired resistance with mere mention of T790M. Articles mentioned above were excluded. Authoritative reviews or chapters were reviewed. Pertinent articles were carefully chosen according to the intention of the review to provide an overview on T790M in patients with lung cancer and the available treatments. The related references of the chosen articles were also retrieved. Characteristics of the studies (patient amounts, distribution of sex and race, sources of the tumor samples, sample amounts, acquired time of the samples, percentage of adenocarcinoma, T790M analytic methods, the presence of T790M and MET in the samples) were extracted from published articles and summarized to aid comparison. These data were extracted independently by two authors. When there were differences in the interpretation, a consensus was often reached after discussion. If not, we re-discussed it with a third author and made the final interpretation.
|
|
Results
22 articles were chosen for analysis (Fig 1).

Fig 1 Inclusion and exclusion flow diagram
|
|
Sample and laboratory approach
Most of the clinical specimen came from original lung cancers or metastatic tumors, while part of the studies examined the DNA extracted from plasma or pleural fluid. The detection methods included PCR/qPCR with or without direct sequencing (DS), amplification refractory mutation system (ARMS) or Scorpion Amplification Refractory Mutation System (SARM).
|
|
Case report
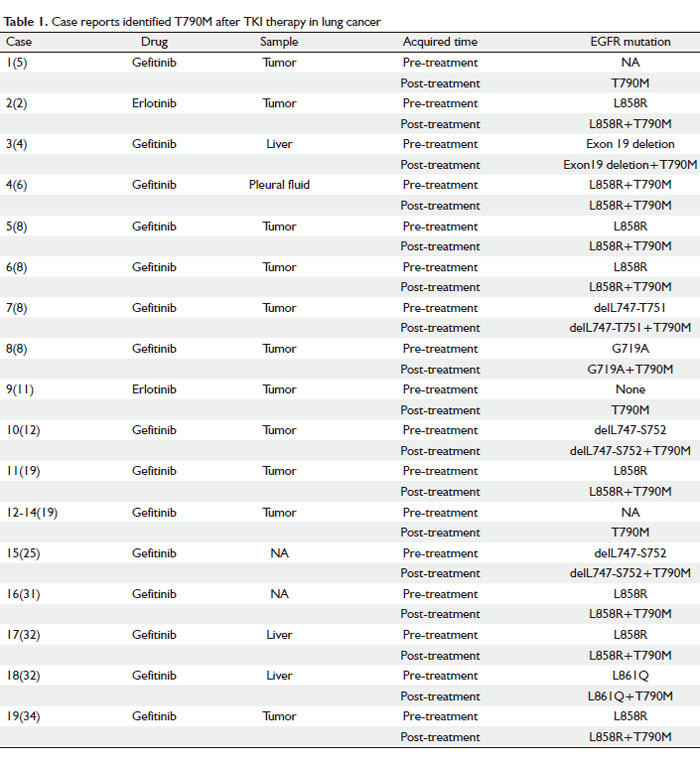
The characteristics of 19 patients from 12 case reports were summarized and listed in Table 1. The EGFR mutational status of most cases was analyzed by DS and PCR techniques. Among the 19 samples, 18 were from the original or metastatic tumors and only one sample was from the pleural fluid (6). Before the TKI therapy, 14 patients were reported to harbor different kinds of EGFR sensitive mutations, such as delL747-S752 of exon 19, L861Q and L858R, which were supposed to respond well to the TKI treatment. Only one of the 19 samples was tested positive for T790M before the TKI therapy (7); however, it became dominant in all 19 patients after the TKI therapy failed. The results indicated that even in the presence of sensitive mutation such as L858R-positive allele, T790M emerged on an L858R-positive allele and produced a T790M-L858R double-mutant allele which can still confer gefitinib resistance. It also showed that characteristic EGFR activating mutations seem to occur preferentially in cis with the T790M mutation.
Cases 5-8 reported four members of a family of European descent in which multiple members developed the bronchioloalveolar carcinoma (BAC) subtype of NSCLC. It is a germline EGFR T790M “drug-resistance” mutation associated with familial NSCLC. The observation this germline EGFR mutation specifically leads to BAC highlights the extreme sensitivity of this cell type to alterations in EGFR signaling, which may explain the high incidence of EGFR mutations in lung cancers with this histology (8).
It was found that after TKI therapy failure, T790M exists in a fraction and not all of the cells within the recurrent tumor. Furthermore, even in different samples from the same patient, secondary T790M is heterogeneous. These observations strongly indicated that additional mechanisms of resistance could be involved in cases with or without T790M mutation (9).
In conclusion, T790M mutation is occasionally present as a minor population in tumor cells before treatment and it is usually concurrent with other EGFR mutations. TKI-resistant tumors are heterogeneous indicating there are other mechanisms involved in the acquired resistance.
 |
|
Clinical report
T790M in TKI-naïve patients
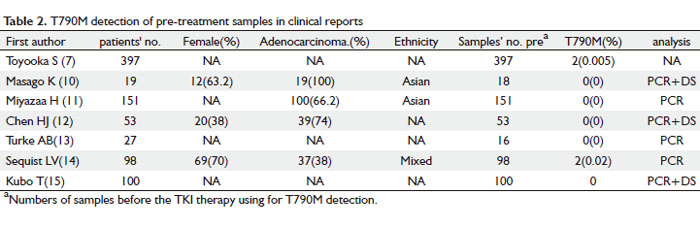
As shown in Table 2, T790M was detected as a large-scale screening in TKI-naïve patients in seven reports. The clinical characteristics of the patients involved are listed in the table 2. Of the 845 patients, only four were tested positive for T790M, which correlated to 0.47% (4/845). This is consistent with previous findings that T790M can exist as a minor allele before TKI treatment. Moreover, all four T790M positive patients were found to have inherited double mutations with a concurrent mutation L858R, which happened to be a very aggressive type of tumors. The patients later had recurrent disease rapidly and eventually died (3,10-15).
T790M in TKI-resistant patients
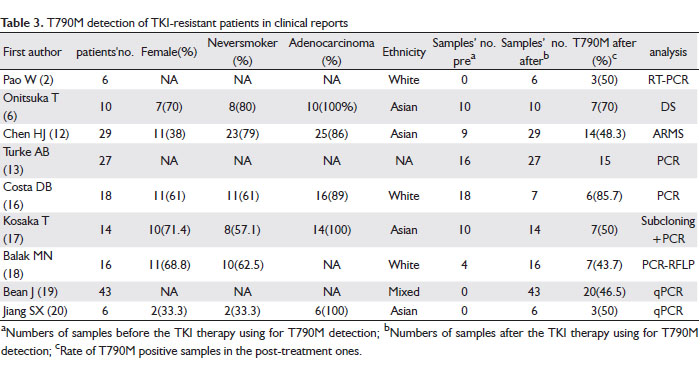
Table 3 is a data summary of 169 TKI-resistant patients obtained from nine clinical reports on TKI acquired-resistant patients. T790M mutation was not found in 67 pre-treatment samples, but 82 of 158 samples were tested positive after TKI therapy failure. In Costa DB’s report (16) the T790M was present at a much higher frequency; however, only seven of those 18 resistant patients were re-sampled. Thus the sample amount was too small to be representative of all patients. The percentage of T790M in TKI acquired-resistant patients in most of the reports are around 50%, with no strong association of sex, smoking-status, pathological subtype, and ethnicity.
Treatment of Gefitinib-resistant and T790M-acquired patients with erlotinib
A question that remains unclear is whether gefitinib-resistant and T790M-acquired patients could benefit from a switch to erlotinib treatment. There are ten articles we collected talking about this kind of cases. (16,21-29).
It was found that majority (>83%) of the gefitinib-resistant patients given erlotinib (150 mg/d) had radiographic progression within the first two to four months of exposure (16,21), and one case report also reported a lack of efficacy from the switch (22). In contrast, some have reported the tumors to shrink and a partial response was achieved with erlotinib treatment (23,24). Similarly, the results from a phase II study show that erlotinib seems to be a potential therapeutic option for treating advanced NSCLC patients with wild-type EGFR who had stable disease while receiving gefitinib (25).
Some debate still exists regarding the efficacy of erlotinib on a highly selected characteristic subgroup (female, Asian, nonsmoker, adenocarcinoma) of advanced NSCLC. While one study reported an unsatisfactory efficacy (26), another reported the patients who responded favorably to gefitinib in the first-line setting, could attain a disease control rate of 66.7% by salvage treatment with erlotinib. Since only stable disease was achieved as the best overall response, this treatment approach should be adopted for patients who had demonstrated good response to previous gefitinib treatment. Otherwise, alternative treatment approach with systematic chemotherapy or best supportive care would be offered (27), which is in agreement with Lee’s phase II study (28) and Vasile’s case reports (29).
T790M and MET amplification in TKI-naïve and TKI-resistant patients
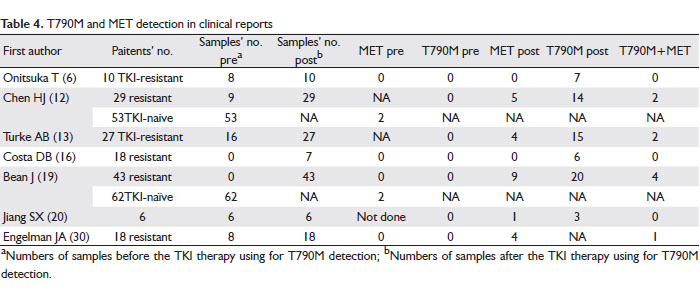
Recently, MET amplification is more and more referred as another important mechanism of TKI resistance. In order to have a better understanding of the relationship between T790M, MET amplification and TKI resistance, we extracted data from seven reports that examined MET amplification in TKI resistant or TKI naïve NSCLC patients and also T790M. The data for 115 TKI-naïve patients, 6 TKI-responsive patients and 145 TKI-resistant patients are summarized and listed in Table 4. For the case studies of TKI-treated patients, 47 samples before TKI therapy and 140 samples after were collected. MET amplification was not found in any of the samples before the treatment, but found in 23 samples after treatment. T790M was detected positive in 66 samples post treatment, but in none of the pretreatment samples. Interestingly, nine patients had concurrent T790M mutation and MET amplification, indicating a high proportion of patients (approximately 39.1% in this literature review) with MET amplification also have T790M. In contrast, MET was amplified in only 3.48% (4/115) of TKI-naïve patients, but highly amplified in 15.9% (23/145) of TKI resistant patients (6,12,13,16,19,20,30).
Based on the 51.9% TKI-resistant patients with T790M from this literature review, we conclude that T790M will continue to be the most prevalent cause of EGFR-TKI resistance. MET amplification with or without T790M, which was previously reported to activate ERBB3 leading to resistance, is also involved in the mechanisms of TKI resistance. This finding may have important implications for TKI-resistant patients. Irreversible EGFR inhibitors, which are currently under clinical development as treatment for patients whose tumors have developed acquired resistance to TKI, may be ineffective in the subset of tumors with a MET amplification even if they contain an EGFR T790M mutation (30).
In general, MET amplification accounts for about 20% of TKI acquired-resistant patients by a different molecular pathway from T790M and part of them will concurrently have T790M mutation. Irreversible TKI might not be effective on these patients.
Comparison of T790M detection methods
In most cases, DS plus PCR were used to detect EGFR mutation status including T790M. Generally, PCR, especially mutant-enriched PCR is a more sensitive and highly specific assay that can detect one copy of mutant allele among as many as 103 to 104 copies of wild-type alleles when compared to direct DNA sequencing. The use of this assay has been validated with various clinical specimens, including biopsy and pleural fluid specimens (31).
The use of Scorpion Amplification Refractory Mutation System (SARM) was also reported, in which the two technologies ARMS and Scorpion were combined to detect T790M in real-time PCR reactions (12). Compared to traditional PCR, SARM system was more sensitive.
In one clinical study, both SARM and WAVE/Surveyor methods were employed for the detection of EGFR activation and resistant mutations using plasma DNA from TKI resistant patients. Eight out of nine T790M positive patients were detected by SARM, while WAVE-Surveyor system detected four, and three were detected by both methods. Whole genome amplification of samples seems to have the greatest effect on the detection of T790M. Furthermore, whole genome amplification of DNA samples was also investigated to determine if the detection of additional EGFR mutations would be facilitated. For EGFR del E746_A750 and L858R, whole genome amplification identified only four additional patients with mutations, whereas for EGFR T790, whole genome amplification resulted in the identification of 10 additional patients (P = 0.011). This may be explained by the fact that T790M often exists as a rare allele and thus may go undetected in the absence of whole genome amplification. The results suggest that whole genome amplification should be incorporated in future noninvasive method in order to aid in monitoring drug resistance and in directing the course of subsequent therapy (32).
In general, PCR, especially mutant PCR is more sensitive than direct sequencing. Since T790M is often present as a minor allele, whole genome amplification could improve detection of T790M.
To sum up, acquired-resistant patients usually undergo a dramatic response or partial response for around 10 months before they develop resistance to TKI. Few patients among them can be detected T790M positive; but after the resistance developed, T790M mutations account for half of these cases. Meanwhile, in our analysis, EGFR T790M always coexist with other resistance mutations such as MET amplification. The switch therapy from gefitinib to erlotinib might be useful for those originally well-responded patients. Irreversible TKIs in development might also show promises in overcoming T790M-induced resistance. The use of high-sensitivity analytical techniques may aid in finding suitable individualized therapy for patients based on their mutational status.
   |
|
Discussion
The first case of EGFR-T790M was discovered in 2005 from a 71 years old male patient (former smoker) with EGFR-mutant (delL747-S752), gefitinib-responsive and advanced non-small-cell lung cancer who had a relapse after two years of complete remission with gefitinib treatment. The DNA sequence of the EGFR gene in his tumor biopsy specimen at relapse revealed the presence of T790M. The T790M mutation has since been identified as a novel, second mutation of the EGFR gene that might lead to resistance to TKI therapy (4). Interestingly, a mutation analogous to T790M was also observed in other kinases with acquired resistance to another kinase inhibitor, imatinib (2).
Currently T790M mutation is believed not only to be a cause of resistance to TKIs, but also an oncogenic mutation that confers growth advantage to cancer cells by increasing phosphorylation levels. However, one report noted that T790M mutation showed no bias for sex, smoking status or histology, but was significantly more frequent in advanced tumors (9/111) than in early-stage tumors (1/169). It shows that T790M mutation is associated with a relatively late phase of NSCLC development when tumor cells exhibit more genomic instability. These findings suggest that EGFR T790M mutation does not play a major causative or proliferative role in tumorigenesis (31). The specific role of T790M in lung cancer development remains unclear and further studies are still needed.
It is still unclear why T790M-acquired patients become resistant to TKI therapy. The C-to-T base-pair change is predicted to change threonine to methionine at position 790 (T790M) in the catalytic pocket of the EGFR tyrosine kinase domain. Structual modeling shows that T790M appears to be critical for the binding of TKIs to EGFR and that it is in juxtaposition to the acetylene side chain of the aniline group. Thus it is often referred to as the “gatekeeper residue”. The T790M mutation leads to steric hindrance of TKIs binding owing to the presence of the bulkier methionie side chain (31). Another possibility is that multiple coexisting mechanisms along with T790M mutation could cause acquired resistance either cooperatively or independently. A recent study suggested that increased internalization of ligand-bound EGFR or EGFR gene amplification by altering downstream molecules such as AKT could be the underlying mechanism leading to acquired gefitinib resistance (17).
After reviewing the results from ten reports on gefitinib-resistant and T790M-acquired patients who had switched to erlotinib treatment, we found that no consensus could be reached. Heterogeneity of the study population might account for the conflicting results. In general, erlotinib failed in most of these cases. This could be because gefitinib-resistant patients predominantly harbor T790M and/or MET amplification, which might be cross-resistant to both EGFR-TKIs. However, some patients did benefit from this radical attempt. One possible explanation is that the presence of heterogeneous malignant clones with different EGFR mutation status may confer differential sensitivity to the two EGFR-TKIs. Some of the acquired gefitinib-resistant clones might be nonresistant or incompletely cross-resistant to erlotinib (28). Also, the standard dosages of 250 mg gefitinib and 150 mg erlotinib are not biologically equivalent., which might be related to drug pharmacokinetics and dosage. Erlotinib is less susceptible than gefitinib to metabolize by the cytochrome-P450 pathway with lower clearance rate, thus able to inhibit the activity of wild type-EGFR at lower concentration than gefitinib (27). Furthermore, several reports suggested that a more highly-selected population such as EGFR wild type patients or previously gefitinib responding patients with selected clinical features might benefit more from the EGFR-TKI switch treatment. However, more case reports and larger randomized clinical trials to test the frequency of these cases are still needed to validate this hypothesis, which would aid in the selection of patients who might still respond to erlotinib after gefitinib treatment failure.
At present, noninvasive genotyping should not replace the gold standard of repeated tumor biopsy, because there are potential limitations to all plasma-based tumor DNA genotyping methods. Firstly, the technique lacks the ability to determine whether the isolated DNA is truly tumor-derived. One potential method to overcome this limitation is to specifically isolate circulating tumor cells and use them for the genotyping studies. Secondly, it would be difficult to determine whether the plasma-derived DNA changes are truly reflective of the genomic changes of the bulk of the cancer. It will be important to correlate the plasma-based studies with tumor-based studies. An additional factor that needs to be considered in noninvasive evaluation of EGFR T790M is the time between the development of resistance and the collection of the plasma DNA specimen for evaluation. Validation of the noninvasive method in prospective clinical trials is necessary to determine its sensitivity and specificity (1). The success of this method has the potential to eliminate the need for repeated tumor biopsies in the detection of T790M mutation. In the future, the incorporation of more sensitive systems, such as those based on high-performance liquid chromatography (HPLC), tandem mass spectrometry or high-density picoliter reactors may also be useful for detecting EGFR T790M (1).
Recently, several kinds of irreversible small molecule TKIs capable of binding covalently to the catalytic pocket of the receptor have received tremendous interests. This is mainly because they also retain activity against tumors harboring some common resistance mechanisms, including T790M. In one model system report, the irreversible EGFR inhibitor compound CL-387, 785 was more effective than gefitinib in inhibiting the growth of H3255 GR cells in which a small proportion of the amplified alleles had acquired a T790M mutation. While both gefitinib and CL-387, 785 induced apoptosis in the gefitinib-sensitive cell line, only CL-387,785 induced apoptosis in H3255 GR cells. Another irreversible TKI, BIBW 2992 also demonstrated more potent activity than gefitinib and erlotinmib in inducing the cell death in cell lines including those with erlotinib-resistant T790M mutations. The similar story happened to HKI-272 (neratinib), which can suppress ligand-induced EGFR autophophorylation and downstream signialing and growth-inhibiting NCI-H1975 BAC cells harbouring T790M mutation. The results of another irreverxible pan-HER TKI, PF00299804 in xenograft models are also promising in anti-tumor activity to overcome T790M mediated resistance (33). These similar preclinical studies all contribute to a growing body of evidence suggesting that irreversible TKIs may be an effective treatment for patients whose tumors harbor the T790M mutation (34).
The biggest limitation of this study is that the articles we collected and analysed are all clinical articles. Due to the limitations of time and knowledge, we do not include those lab researches of T790M, thus the results on the animal models are excluded from this review, which might compromise the comprehensive knowledge of T790M current researches. Meanwhile, the clinical reports about T790M are still very limited and variegated; we still cannot address some of our objectives clearly such as which kind of T790M-acquired patients will benefit most from switch therapy. It still needs larger randomized, controlled clinical trials to find out and confirm.
|
|
Conclusions
T790M mutations are found in about half of the TKI acquired-resistant patients while in few TKI-naive patients. The use of high-sensitivity analytical techniques may aid in finding suitable individualized therapy for patients based on their mutational status. T790M-harbored patients may benefit from switch therapy or irreversible TKIs. Further prospective randomized studies would be valuable in determining the association between T790M and lung cancer tumorigenesis and prognosis, the characteristics of potential patients who might benefit from switch therapy and discovering new corresponding treatment to overcome the acquired resistance.
|
|
References
Cite this article as: Ma C, Wei S, Song Y. T790M and acquired resistance of EGFR TKI: a literature review of clinical reports. J Thorac Dis 2011;3(1):10-18. doi: 10.3978/j.issn.2072-1439.2010.12.02
|

